
Regulatory affairs (RA) teams are under pressure more than ever before. Faced with a growing demand for pharmaceuticals, cumulative legislative changes and an approaching steep patent cliff, sponsors are struggling to find new ways to modernize, innovate and deliver therapeutics to market faster.
The greatest challenge isn’t just the volume of work, but the unpredictable complexity of a global regulatory landscape. Unlike demographic shifts such as the doubling of the world’s population aged 60 and above by 2050, or the $350 billion worldwide revenue loss that the industry will support through 2030 due to loss of exclusivity (LoE), regulatory change is a moving target, influenced by public health policies, technological progress and political agenda.
Legal and regulatory changes: growing pressure on the pharma industry
Looking ahead, the FDA and EMA will continue to push for modernization, introducing new frameworks and guidance around digitized submissions and AI. The new regulations demand that companies demonstrate their capabilities in areas such as:
- Electronic submissions
- AI model validation
- Enhanced clinical data transparency
The goal to accelerate review timelines and enhance data integrity is evident. However, these changes also put additional pressure on sponsors, who need to revise submission formats and workflows, upgrade their technology platforms and retrain their regulatory teams to meet the latest requirements.
At the same time, regulatory reforms like the U.S. Inflation Reduction Act and revisions to the European Union’s pharmaceutical legislation are pressuring profit margins and altering decisions about strategic research and development (R&D) allocations. Industry experts predict that the Inflation Reduction Act to drive a 31% decrease in US pharma revenues through 2039 and longer approval timelines for new therapeutics.
This perfect storm has made one thing clear: optimizing submission workflows and expediting regulatory filings is a matter of strategic urgency.
“[…] biopharma leaders will have to rethink their go-to-market strategies to effectively address these access challenges and adapt to shorter product lifecycles.”
Qasim Zaidi – Managing Director, Health Sciences & Wellness, Ernst & Young LLP, September 2025
Source: Driving growth via commercial transformation in pharma | EY – US
Regulatory change management: best practices
Regulatory change is constant and frequent. The change management process may be somewhat similar every time, although this does not necessarily mean smooth sailing. Once regulatory intelligence has been centralized, RA teams proceed to prioritize the implementation, based on factors such as:
- Relevance to therapeutic area or target molecules
- Impact on clinical trials (ongoing and planned)
- Submission process disruption
At this stage, a gap analysis is performed to identify the current state and the ideal future state that organizations should aim to achieve.
In this article series, we will explore the current state and ideal future state that sponsors should consider when optimizing their regulatory submission practices. But before we delve into that analysis, let’s quickly define the key performance indicators (KPIs) relevant to sponsors for measuring the efficiency of regulatory submissions management.
Measuring up: the KPIs of regulatory efficiency
Regulatory affairs have a set of well-defined KPIs that the industry has developed over the years to ensure productivity and timeliness are met. Among a vast array of KPIs that pharma organizations track, here is an essential, non-exhaustive list that regulatory teams follow to ensure high regulatory submission productivity and efficiency. These metrics offer a view of where key bottlenecks lie:
1. Time-to-market represents the entire duration from early-stage drug discovery or lead identification through to commercial launch. Companies that can bring drugs to market faster gain a competitive and financial edge.
Identify: inefficiencies or bottlenecks in research, clinical development and regulatory submission processes
By monitoring you can: reduce costs, accelerate approvals and deliver new treatments to patients more quickly
2. R&D cycle time metrics is a metric you can use to track specific milestones throughout drug development, e.g. how long each clinical phase takes on average, or the elapsed time from selecting a drug candidate to submitting an IND application.
Identify: inefficiencies at different stages, from preclinical work through trial execution and regulatory filing
By monitoring you can: create a more responsive and streamlined development process
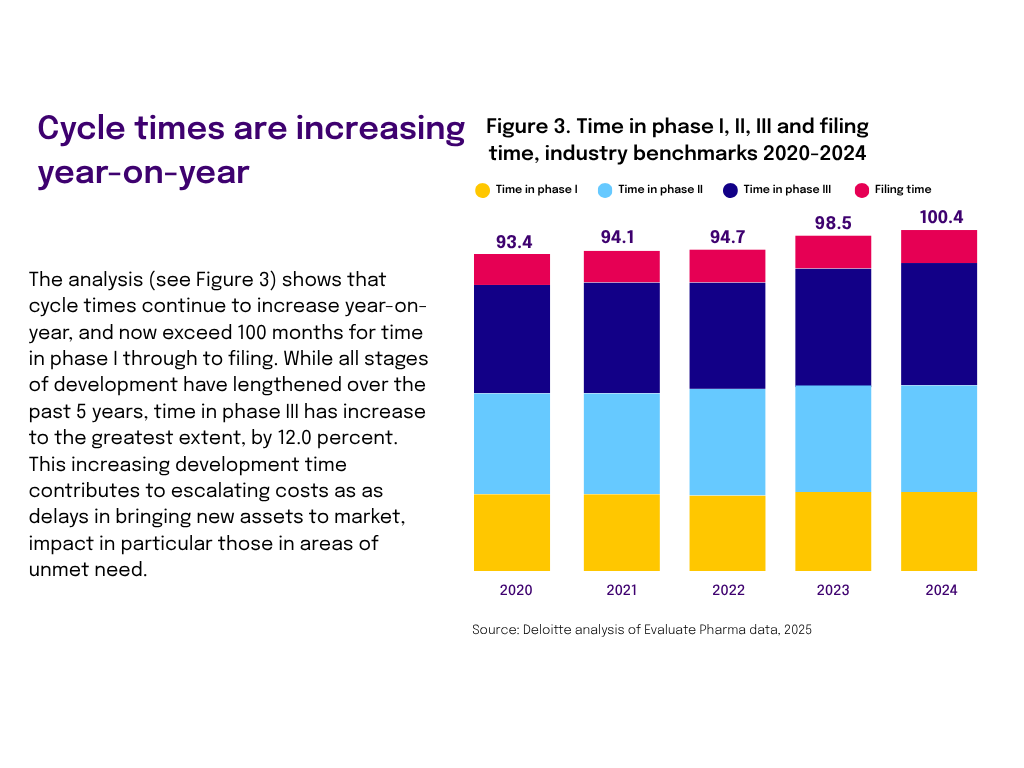
3. R&D cost per new drug measures how much a company spends on research and development for each drug that ultimately secures regulatory approval. A lower cost per approval typically signals stronger portfolio selection, better-designed trials, and tighter operational execution.
Identify: excess attrition or process inefficiencies
By monitoring you can: reduce costs
4. Documentation and Record Compliance are a set of key metrics you can track to see whether documents stay current (for example, ensuring no SOPs are overdue for review) and whether data integrity standards are maintained. Following several notable regulatory violations, data integrity has become a critical priority. The objective is to minimize documentation errors and data integrity incidents as much as possible.
Identify: if documents are up to date, if data integrity standards are maintained
By monitoring you can: reduce costs, expedite submission timelines
5. Regulatory submission timeliness KPIs focus on submitting dossiers, e.g., INDs, NDAs, and variations, by or before their scheduled deadlines. Equally important is the speed of responding to questions from health authorities during their review process. Timely submission and rapid query resolution are vital for preventing approval delays.
Identify: how fast your organization is responding to regulatory authority queries
By monitoring you can: avoid approval delays, achieve timely regulatory submissions and regulatory approval timelines
6. Audit findings and inspection outcomes are monitored to capture both the count and gravity of issues uncovered during regulatory inspections (for example, FDA Form 483 observations or EMA GMP inspection deficiencies) as well as internal quality audit results. The objective is to steadily decrease overall findings and eliminate any critical nonconformances. Monitoring year-over-year changes, such as a drop in recurring observations, provides a clear indicator of strengthening compliance controls.
Identify: speed of responding to regulatory authority queries, timely submissions times
By monitoring you can: avoid approval delays, reach proposed regulatory submissions and regulatory approval timelines
Excerpt A: “The Manufacturing Scientific Analytical Technology (MSAT) laboratory used in support of application submission testing data had inadequate controls over data integrity.”
Excerpt B: “There is a failure to thoroughly review any unexplained discrepancy whether or not the batch has been already distributed.”
Excerpt C: “Laboratory records do not include complete data derived from all tests, examinations and assay necessary to assure compliance with established specifications and standards.”
Excerpt D: “Laboratory records are deficient in that they do not include a complete record of all data obtained during testing.”
Excerpt E: “Written records of investigations into the failure of a batch or any of its components to meet specifications do not always include the conclusions and follow-up.”
Excerpt F: “All your required records do not include the time of the activity that the records reflect.”
Excerpts from FDA 483 Forms
Source: FDA Dashboards – Inspections
Now that we’ve seen the main KPIs that sponsors or CROs are monitoring to strengthen their capabilities in producing regulatory content, let’s take a closer look at the current challenges they are facing.
The current state: internal and external challenges
Many organizations are struggling with chronic challenges from both internal and external factors.
1. Volume overload and resource strain
Organizations are experiencing a significant increase in regulatory submissions due to growing pharmaceutical pipelines, lifecycle management efforts, and expanding global markets. Companies are handling dozens, if not hundreds, of submissions annually across different product types, NDAs, INDs, CTDs, variations and post-approval changes. This surge is driven by the rapid development of new therapies, especially personalized medicines, biosimilars and complex biologics, pushing regulators to process more dossiers to meet market demands.
Key challenges:
- Human resources strains
- Prolonged submission timelines
- Quality risks (increased likelihood of mistakes in documentation or formatting)
- Managers struggle to prioritize submissions amidst the sheer volume, risking overlooked deadlines or incomplete dossiers due to managers not being able to correctly prioritize workload for teams.
2. Complexity from multi-country regulations and evolving standards
Global pharmaceutical companies operate in an environment with highly fragmented regulatory landscapes. Each country or region — e.g., the U.S., EU, China, Japan — has its own requirements regarding dossier formats, data standards, clinical trial regulations, and review procedures. These requirements are continually evolving with new scientific and policy developments, for example, updates to AI validation guidelines or real-world evidence standards.
Key Challenges:
- A vast array of formats and languages
- Frequent updates and revision demands
- Cross-border submissions that need to meet the specific demands of each jurisdiction
- Failure to adapt promptly can lead to rejection, delays, or additional queries
3. Data silos and resource constraints
Essential data for regulatory submissions (clinical trial data, manufacturing records, pharmacovigilance reports) is often stored in disjointed systems or silos. Siloed systems hinder seamless data flow, making it difficult for regulatory teams to access comprehensive, up-to-date information during dossier preparation.
Key Challenges:
- Fragmented systems that lead to delays in gathering and validating data
- Time-consuming and error-prone manual data consolidation
- Interdisciplinary collaboration hampered by disjointed data hampers
- Data harmonization and integration affected by limited staff and technological capacity
4. Challenges of traditional manual and document-intensive submission processes
Many organizations still rely heavily on traditional workflows involving Word documents, PDFs, and Excel spreadsheets for dossier preparation. These manual processes are laborious, with multiple rounds of review, version control issues, and a high likelihood of human error. This approach is incompatible with the speed and complexity of today’s regulatory environment.
Key Challenges:
- Time-consuming processes
- Version control and errors that lead to compliance risks
- Scalability issues
5. Risks associated with errors, inconsistencies and regulatory non-compliance
Errors and inconsistencies, such as incorrect data, formatting issues, or missing documentation, remain prevalent within submission dossiers, primarily due to manual processes. These issues can lead to operational delays, extra rounds of review or inspection findings. Among key regulations, directives and good practices that pharma needs to align with, for successful regulatory submissions in the US and the EU, we can mention:
- U.S. FDA 21 CFR Part 11 (Electronic Records and Signatures) mandates controls for electronic data integrity, audit trails and secure user authentication.
- U.S. FDA 21 CFR Part 314 (Applications for FDA Approval to Market a New Drug) specifies dossier format, content requirements and post-market reporting obligations for NDAs and ANDAs.
- EU GMP Annex 11 (Computerized Systems) outlines data integrity controls, validation and audit‐trail expectations for electronic submission packages.
- EU Directive 2001/83/EC & Regulation (EC) No 726/2004 establishes marketing-authorization procedures, dossier standards (including the CTD structure) and pharmacovigilance requirements.
- GDPR (General Data Protection Regulation) must be applied when clinical-trial data includes personally identifiable information for EU subjects.
- ICH M2 defines the structure and transmission format to facilitate electronic communication between sponsors and regulators
- eCTD v4.0 is the latest version of the eCTD dossier format, which introduces new XML standards and validation rules beyond ICH M2.
- ICH M4 is a guideline that presents the agreed-upon format of a common technical document (CTD), harmonizing dossier modules across ICH regions, ensuring consistent structure and content for Quality, Safety and Efficacy information.
- ICH M11 establishes the technical specifications for drafting study protocols, enabling machine-readable, semantically consistent documents that streamline protocol authoring, review, and multi-region trial starts.
- ICH Q10 outlines a comprehensive quality management framework covering the entire product lifecycle, promoting continuous improvement and risk management in submissions.
Key Challenges:
- Increased risk of deficiency letters or inspection citations due to incomplete or incorrect dossiers
- Delayed product launches due to re-submissions and corrections
- Reputation damage, sanctions, fines or even loss of regulatory trust
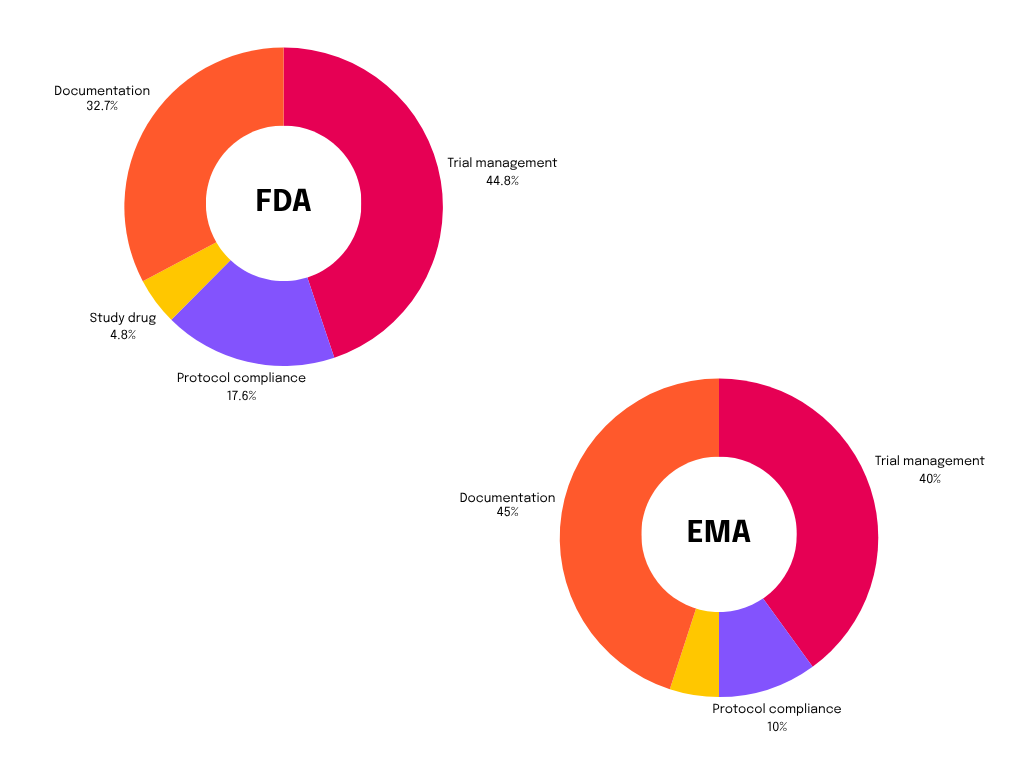
Percentages of good clinical practice findings by deficiency area in 23 Sponsor/Contract Research Organization Inspections for FDA and EMA / Source: Descriptive Analysis of Good Clinical Practice Inspection Findings from U.S. Food and Drug Administration and European Medicines Agency – PubMed
6. Keeping up with regulatory changes
Regulatory agencies continuously update their guidelines, review processes, and data standards, especially in response to new science and digital advances. Staying ahead demands constant monitoring of regulatory news, interpreting complex revisions and adapting internal processes swiftly.
Key challenges:
- Tracking regulatory updates across multiple jurisdictions
- Updating SOPs, training staff and modifying submission templates
- Maintaining uniform compliance practices across departments, while at the same time integrating new requirements
- Last-minute changes that cause delays or non-compliance
7. The impact of delays on patient access and company competitiveness
Regulatory delays, often stemming from the mentioned challenges, directly impact patient access to novel therapies, especially in life-threatening conditions or unmet needs. At the same time, delays erode a company’s competitive advantage, leading to lost revenue and market share.
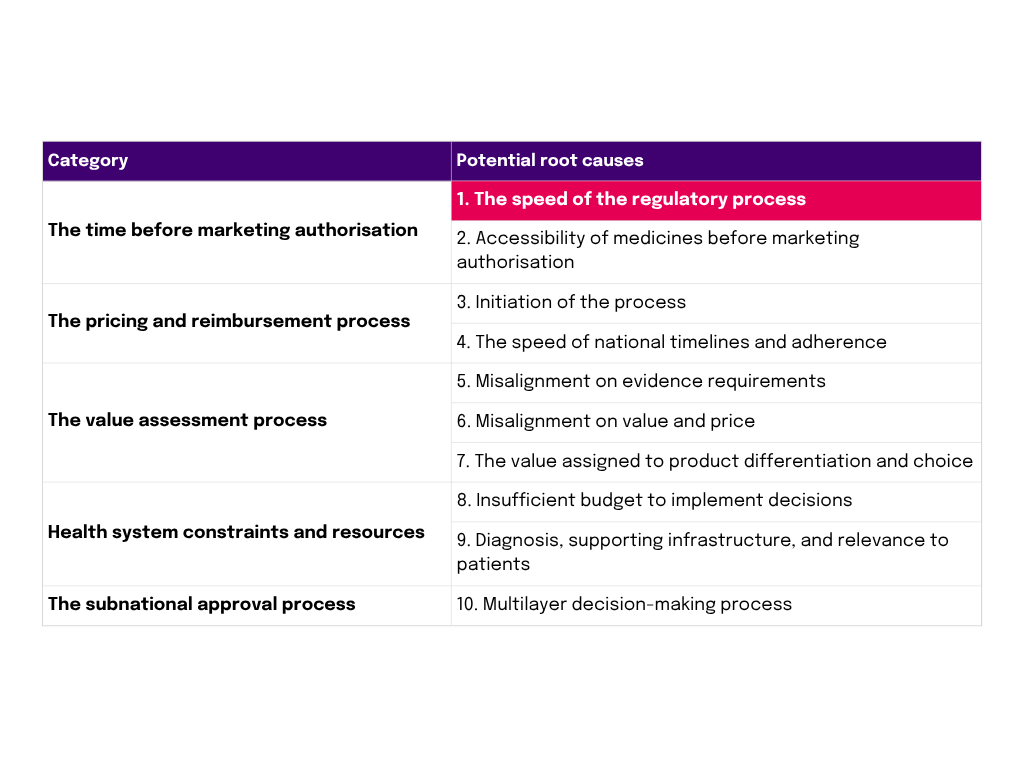
The root causes of delays and unavailability / Source: The root causes of unavailability of innovative medicines and delay in access: Shortening the wait
Looking ahead: the future of regulatory submissions optimization
The evidence is clear: pharma organizations need new strategies and tools to overcome the current technological, regulatory or macroeconomic conditions, along with the associated challenges. The solution is not to work harder but to fundamentally assess the information architecture underpinning the way documentation is created, managed and submitted and the outcomes they can achieve.
Stay tuned for the second part of this article, where we will explore the ideal state sponsors and CROs can target to optimize regulatory submissions and mitigate risks and challenges. We will provide insights into the key focus points for an efficient gap analysis and we will discuss the best documentation processes that can help your organization overcome regulatory bottlenecks.
In the meantime, we encourage you to explore our pharma-dedicated blog section. If you don’t want to wait for that second article to find out what’s the latest on how you can move faster, we would be more than happy to meet and discuss your context. We are ready to support your growth.

Product marketeer for Tridion and Fonto One at RWS