
Pharma organizations can spend as much as $4.54B to bring a single molecule to market. The costs are only rising, while regulations are becoming more complex. This pressures organizations to manage extensive portfolios and afferent regulatory dossiers with fewer resources.
When regulatory submissions take too long or are not accepted, organizations incur significant costs, from direct and tangible ones like financial costs of rework and resubmission, delayed market entry and lost revenue, increased operational costs, to more indirect costs incurred by placing the company at a competitive disadvantage, opportunity costs or, in more dramatic cases, reputation loss as a result of non-compliance and legal penalties.
What value can pharma companies unlock through optimized regulatory submission processes?
We’ve discussed potential risks and losses, but what benefits can pharma companies enjoy when regulatory teams increase efficiency? A recent article published by McKinsey & Company states that faster and higher-quality submissions in the drug development phase can bring several advantages to organizations and end beneficiaries alike. While companies can reduce risk, delays and costs, patients can gain faster access to life-saving therapies.
A pharma company making an improved submission for a $1 billion asset one month more quickly than typical would unlock roughly $60 million in net present value (NPV).”.
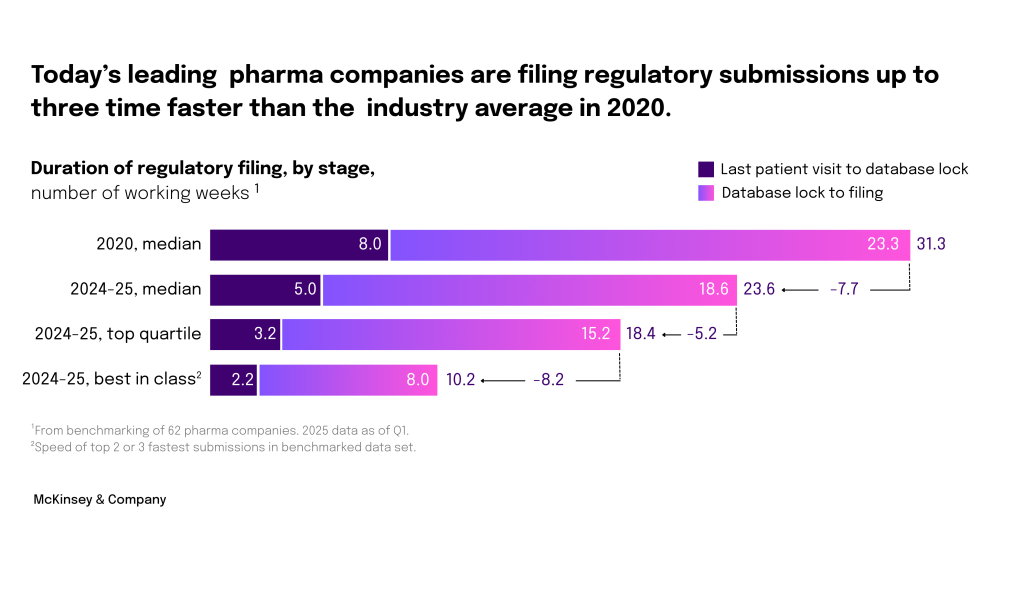
Pharma leaders are advancing continuous improvement in regulatory submissions
Not too long ago, it would have taken a pharma company between 9 months and 2 years, after the last piece of data came in from clinical trials, to submit the dossier to regulatory authorities. Over the past five years, leading industry organizations have achieved a threefold increase in the speed of their regulatory submissions. Top performers are achieving a 10-week interval, aiming to reduce it even further to 8 weeks.
What are the challenges of creating regulatory files?
Nevertheless, most pharma organizations continue to face persistent challenges in shortening submission timelines due to the complexity of regulatory dossiers, difficulties in integrating digital systems, limited understanding of advanced documentation practices, and a general reluctance toward adopting new technologies and moving away from old ways of working.
Inefficient document lifecycle management tools and systems. Most organizations use basic file-sharing platforms such as Word, Excel, SharePoint, Google Drive, or Dropbox, which lack proper version control, audit trails and workflow automation. These tools can’t offer the robust controls required for regulated environments, and using them leads to document and information duplication, inaccurate data and content, version confusion and high compliance risk. Not to mention that these very popular platforms are continuously exposed to cyber-attacks, some of which occasionally succeed, leading to your data being leaked.
Modular point solutions created for specific tasks. Various solutions are utilized for specific needs and lifecycle phases: clinical data collection and management systems, document authoring, Quality Management Systems (QMS), Regulatory Information Systems (RIM) for managing submissions, and project management tools, to name just a few. These do not integrate well, most of the time offering a difficult experience for the people working with them and causing traceability issues, collaboration bottlenecks, difficulty maintaining data integrity and finally lack of visibility across the document lifecycle.
Best practices for effective regulatory dossier compilation
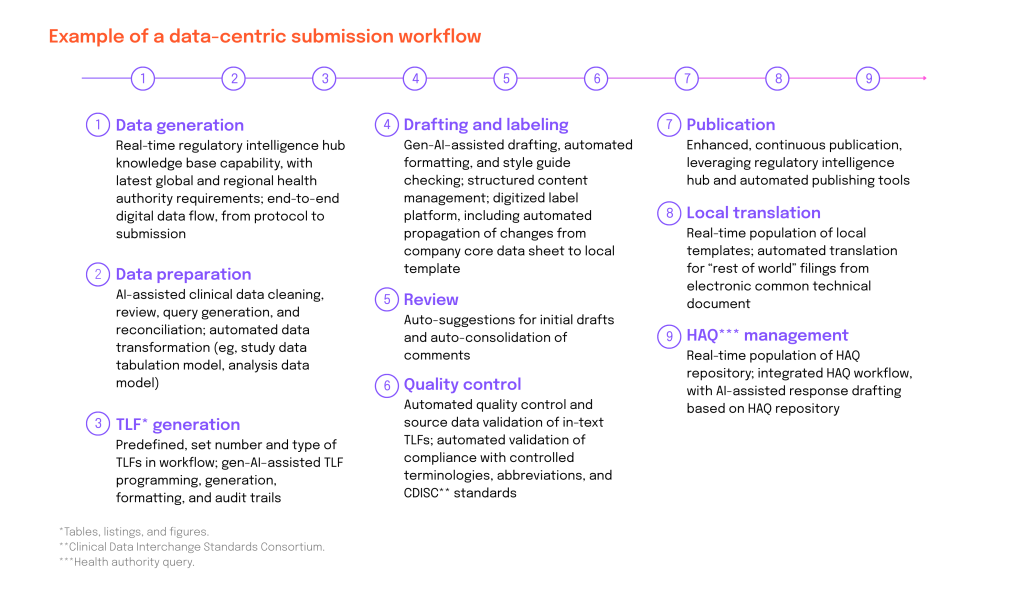
To truly transform regulatory dossier compilation, pharma organizations should embrace a data-centric approach—treating documents as dynamic data assets rather than static files. Three key capabilities can support and enable that evolution:
- Use structured content authoring tools to improve content governance and streamline processes and enable authors, reviewers and SMEs to work collaboratively in real-time.
- Create automation workflows to let editors see when source content or data is updated, so they can evaluate whether the new content version needs to be propagated through the various templates they are managing.
- Enable AI to significantly slash authoring, review and approval times by creating first drafts faster.
Source: McKinsey & Company
Prepopulated templates – a key benefit of structured content authoring
Structured content authoring (SCA) enables medical writers to use prepopulated or dynamic templates by breaking down complex documents into modular, reusable content components that are managed and stored in a centralized system. Using this approach, authors can prepopulate templates with already approved content chunks, e.g. protocol titles, protocol description, tables, listing, figures or other, eliminating time wasted looking for the right information and inserting it into the current working template.
With SCA, medical writers can concentrate on analysis and interpretation specific to the study, rather than duplicating routine content.
With prepopulated templates, medical writers can move faster through the completion of a regulatory document as they can:
- Pull content from the original source for data tables, figure legends, and standard statistical methodology descriptions.
- Maintain consistency across multiregional submissions while allowing for region-specific adaptations.
- Integrate new data into existing document structures for studies quickly, eliminating the need for manual content changes.
- Shorten authoring and review cycles, as writers can choose to automatically propagate pre-approved content across the documentation, without needing to go through re-validation.
- Skip verifying regulatory-required pre-defined content such as disclaimers or other standard mandatory content pieces.
Let’s see how this works and what it looks like in a structured content authoring tool.
Prepopulated templates in Fonto One or Smart Fields
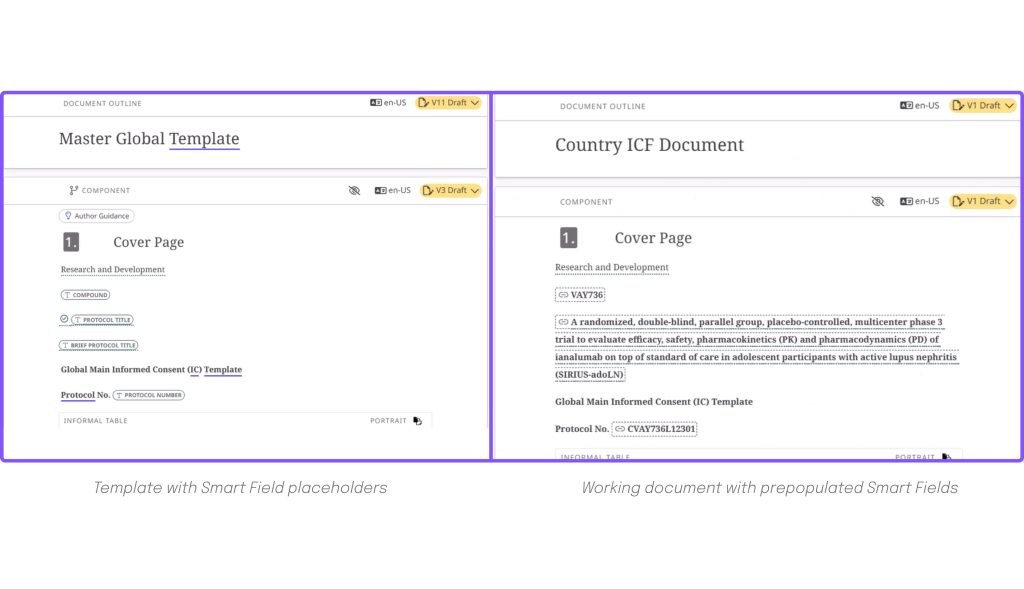
Let’s take an ICF template, for example.
In the first image, you can see the template creator or manager can add to the template what we call Smart Fields in Fonto One: “compound”, “protocol title”, “brief protocol title”, “protocol number”.
When an author starts to work on creating a specific ICF document, they will directly see and work with what is on the right-hand side: a prepopulated document, where the vetted information has been automatically added from the source document. The author can concentrate on valuable content, knowing that all prepopulated information is pre-approved and usable without further review.
Want to learn more about prepopulated templates, structured content and faster regulatory submissions?
From large pharmaceutical and biopharma companies, mid-sized and specialty pharma, to generics manufacturers and CROs, structured content and prepopulated templates can considerably reduce regulatory submission filing times.
Explore how medical writers, reviewers and SMEs can reduce time-to-filing by using accurate, validated prepopulated templates with Fonto One.

Product marketeer for Tridion and Fonto One at RWS